Method
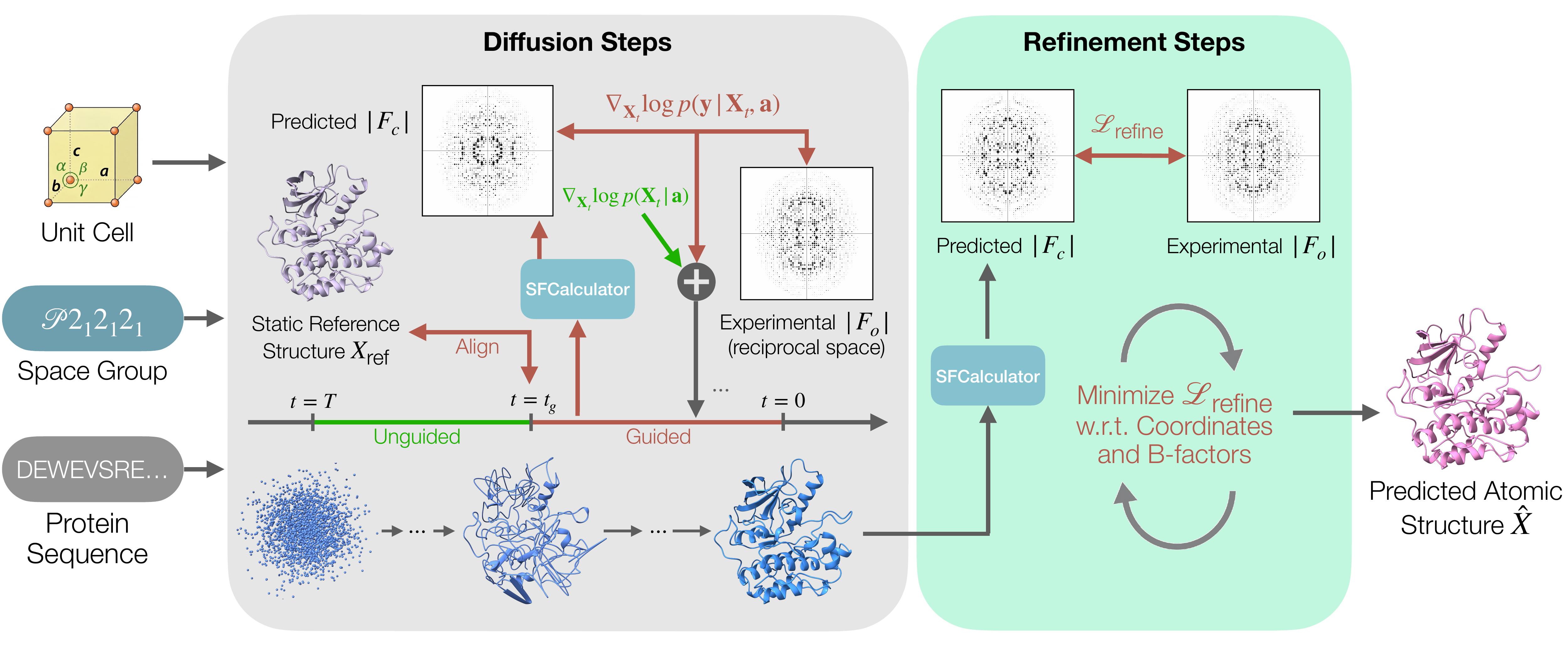
CrystalBoltz proceeds in two phases. Phase 1 runs Boltz-2 reverse diffusion conditioned on the protein sequence together with the crystal's unit cell and space group. Sampling begins unguided to let the prior establish a coarse fold; once a backbone has emerged (at step tg), we switch on experimental guidance and continue through to t = 0. At each guided step, the denoised structure prediction X̂0 is aligned to the crystal frame, passed through the differentiable SFCalculator forward model to produce calculated structure-factor amplitudes |Fc|, and compared against the experimental |Fo| using a combined Gaussian + Rice crystallographic likelihood. The likelihood gradient is backpropagated through the denoiser to steer the sampling trajectory toward structures consistent with the diffraction data. Phase 2 takes the final denoised structure and jointly refines atomic coordinates and isotropic B-factors against the crystallographic R-factor for a small number of Adam steps. This separates global, prior-guided conformational search from local crystallographic fitting.